Research
Welcome to the Werner lab!
We study the molecular and cellular mechanisms of tissue repair, with emphasis on the roles of growth factors and their downstream targets in this process. We are particularly interested in the parallels between wound healing and cancer at the cellular and molecular level.
Injury to adult tissues initiates a series of events, which finally lead to at least partial reconstruction of the injured body site. Except of the liver, which can completely regenerate following acute and moderate injury, repair of other organs is imperfect and results in scar formation with functional impairments. There are many conditions in humans, which are associated with impaired tissue repair, including old age, steroid treatment and several diseases, such as diabetes and cancer. Therefore, there is a strong need to improve the healing process. This requires a detailed understanding of the underlying cellular and molecular mechanisms. By trying to elucidate these mechanisms, our research shall help to develop new strategies for the improvement of tissue repair, in particular of cutaneous wounds. To achieve these goals, we use state-of-the art approaches, including functional genomics, proteomics, and metabolomics, organotypic cell culture systems, mouse genetics, and analysis of human tissue samples.
An exciting aspect of our research is the analysis of the parallels between tissue repair and cancer at the molecular and cellular levels. We identify and functionally characterize genes and signaling pathways, which orchestrate both processes, with a focus on the role of growth factors and transcriptional regulators in tissue repair and cancer. We use the mouse as a model organism to address these questions. Collaboration with clinical partners helps to determine the importance of our findings for the human situation and to transfer our research results into clinical practice.
Cited References from our laboratory can be found under "Publications".
Project 1:
Fibroblast growth factors and activin in tissue homeostasis, wound repair, inflammatory skin disease and cancer (currently funded by ETH Zürich, Swiss National Science Foundation, Wilhelm-Sander Foundation, Cancer Research Switzerland, The LOOP Zurich); previously funded by Roche Foundation, Studienstiftung des Deutschen Volkes, Deutsche Forschungsgemeinschaft, Boehringer Ingelheim Fonds, Janggen-Pöhn Foundation, Schering Foundation, China Scholarship Council, Swiss Government).
Fibroblast growth factors
Fibroblast growth factors (FGFs) comprise a family of 22 proteins, which play important roles in development, tissue homeostasis, repair and disease. We are particularly interested in FGF7, which is also called keratinocyte growth factor (KGF) (Werner, 1998). FGF7 is a secreted protein, which is produced by various types of mesenchymal cells and by gd T cells, but not by epithelial cells. However, epithelial cells express FGFR2b, the only known high-affinity receptor for FGF7. FGF7 is weakly expressed in normal skin but strikingly upregulated in dermal fibroblasts after skin injury (Werner et al., 1992). FGFR2b is expressed on keratinocytes of the epidermis and the hair follicles in mice and humans, suggesting that FGF7 stimulates wound reepithelialization in a paracrine manner. This hypothesis was supported by the delayed wound healing in transgenic mice expressing a dominant-negative Fgfr2b mutant in the basal keratinocytes of the epidermis (Werner et al., 1994), and in mice lacking Fgfr1 and Fgfr2 in keratinocytes (Meyer et al., 2012). Mechanistically, this was caused by impaired migration of Fgfr1/Fgfr2-deficient keratinocytes due to reduced expression of focal adhesion proteins (Meyer/Müller et al., 2012; Fuhr/Meyer et al., 2015).
FGFs are also important regulators of skin morphogenesis and homeostasis. Thus, mice lacking Fgfr1 and Fgfr2 in keratinocytes show cutaneous inflammation, keratinocyte hyperproliferation and acanthosis with strong similarities to Atopic Dermatitis in humans (Yang/Meyer et al., 2010; Sulcova et al., 2015; Seltmann/Meyer et al., 2018: Ferrarese/Koch et al., 2024). We identified loss of FGF-induced expression of tight junction components with subsequent deficits in epidermal barrier function as well as a direct anti-inflammatory effect of FGF7 that is mediated via keratinocytes as the mechanisms underlying the progressive inflammatory skin disease in these mice. This is relevant for human disease, because we found a strong down-regulation of FGFR2 expression in the epidermis of lesional skin of Atopic Dermatitis patients (Ferrarese/Koch et al., 2024). Since Atopic Dermatitis symptoms frequently aggravate at low environmental humidity, e.g. during the dry winter season, we exposed the Fgfr1/Fgfr2 knockout mice that develop Atopic Dermatitis-like symptoms to higher humidity. Remarkably, even a short-term maintenance of the mice at 70% humidity rescued the inflammatory phenotype. Using a combination of quantitative proteomics and functional cell biology we identified the mechanisms underlying the response of the skin to low environmental humidity and discovered the cell adhesion protein CLCA2 as an osmo-regulated protein in keratinocytes that helps to preserve epidermal integrity under stress conditions (Seltmann/Meyer et al., 2018; Seltmann et al., 2024).
In our recent work, we discovered an unexpected antagonism between FGF and interferon signaling. Thus, FGF receptor activation in epithelial cells strongly suppressed the interferon response, resulting in a higher susceptibility to infection with different viruses. Vice versa, FGF receptor inhibition had potent antiviral activities. These results suggest the use of FGF receptor antagonists for the treatment of viral infections (Maddalunod/Urwyler/Rauschendorfer et al., 2020; Stefanova et al., 2024).
FGF7 also exerts potent cytoprotective activities. It protects intestinal epithelial cells from cell death induced by radiation and chemotherapy, and it has been approved for the treatment of mucositis in cancer patients. We confirmed the cytoprotective effect of FGF7 for the skin, and we demonstrated that FGF7 protects keratinocytes from UV- or toxin-induced cell death by stimulating the expression of various genes involved in the control of the cellular redox homeostasis (Braun et al., 2006). Of particular importance is the Nrf2 transcription factor, which we characterize regarding its functions in tissue repair, inflammatory disease and cancer (see project 2).
In analogy to our findings on FGFs in wound healing, we also found a crucial role of FGFR signaling in liver regeneration and in the pathogenesis of liver fibrosis and cirrhosis. Specifically, loss of Fgfr1 and Fgfr2 in hepatocytes caused a severe deficiency in compound detoxification in the regenerating liver through regulation of circadian transcription factors that control various detoxifying enzymes (Böhm et al., 2010). In addition, Fgfr1/Fgfr2 deficiency in hepatocytes suppressed the expression of the epigenetic regulator Uhrf2, which we identified as a potent suppressor of cholesterol and bile acid synthesis. Loss of Uhrf2 in hepatocytes of mice resulted in the accumulation of toxic bile acids in the liver, which induced liver necrosis after partial hepatectomy (Slabber et al., 2023). Combined loss of Fgfr1, Fgfr2 and Fgfr4 in hepatocytes resulted in liver failure after partial hepatectomy, demonstrating that FGFR signaling is essential for liver regeneration (Padrissa-Altes et al., 2015). Additional projects on liver regeneration revealed that b1-integrin and the ubiquitin ligase Nedd4-1 regulate growth factor signaling in the regenerating liver and thereby strongly contribute to the regeneration process (Speicher et al., 2014; Bachofner/Speicher et al., 2017).
Activin
Activins are members the TGF-b superfamily of growth and differentiation factors, which influence proliferation and differentiation of many different cell types. We demonstrated important roles of activin in skin homeostasis and repair. Activin A expression strongly increases in response to skin wounding (Hubner et al., 1996). This is functionally important, since overexpression of activin A in basal keratinocytes of the epidermis of transgenic mice strongly accelerated wound reepithelialization and granulation tissue formation (Munz et al., 1999). The healing-promoting effect is mediated by different types of immune cells and in particular by fibroblasts (Haertel et al., 2018; Wietecha/Pensalfini et al., 2020). However, we also found enhanced scarring in these mice, which resulted from activin-induced expression of pro-fibrotic genes (Wietecha/Pensalfini et al., 2020). Vice versa, inhibition of activin action during wound healing by overexpression of its secreted antagonist follistatin strongly delayed the wound healing process (Wankell et al., 2001).
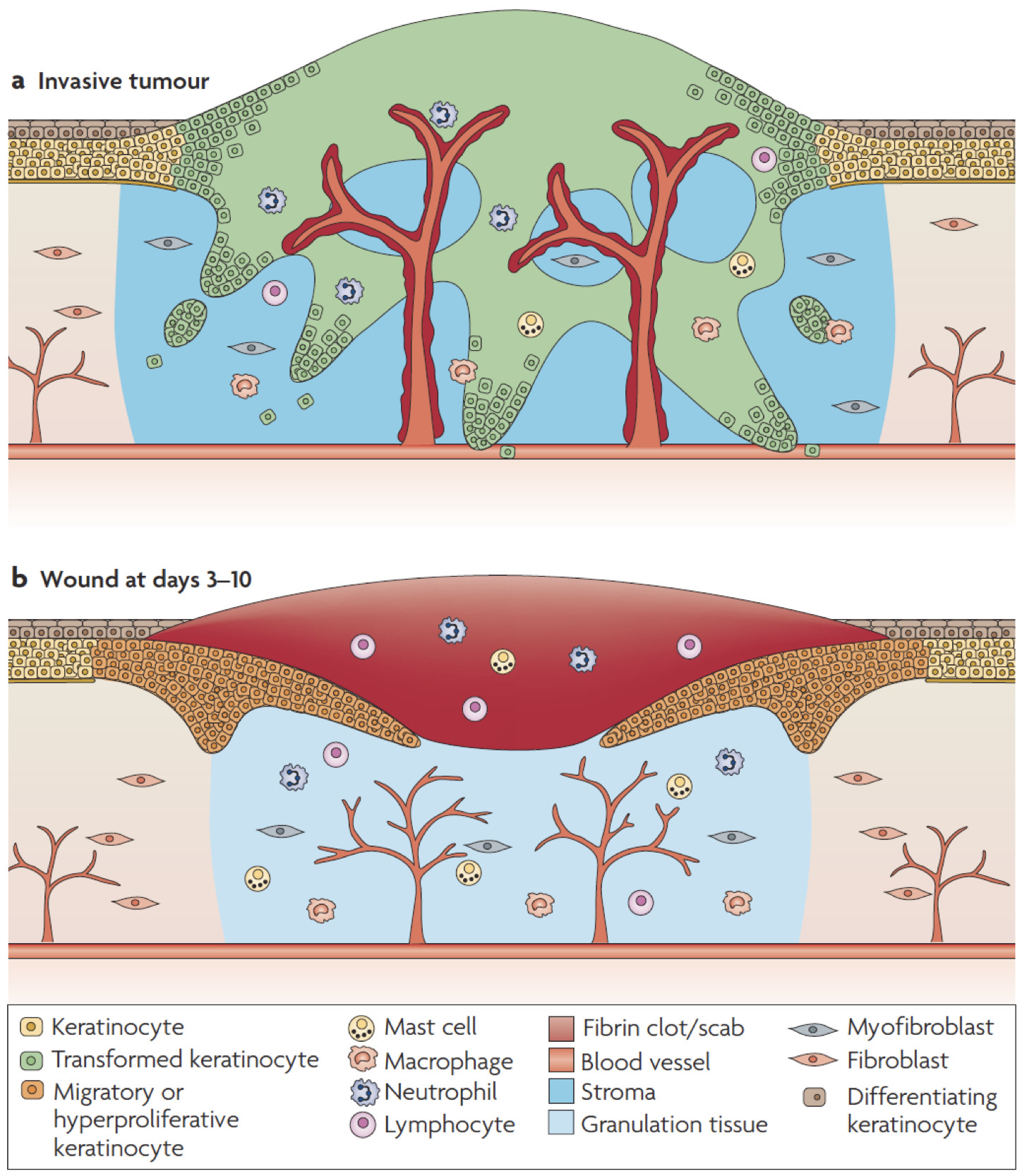
Due to the parallels between wound healing and cancer (Schäfer and Werner, 2008), we speculated about a role of activin in the pathogenesis of skin cancer. Using different types of genetically modified mice, we showed that enhanced levels of activin A in the skin promote skin tumor formation and their malignant progression through induction of a pro-tumorigenic microenvironment. This included accumulation of tumor-promoting Langerhans cells and regulatory T cells in the epidermis and of mast cells in the dermis (Antsiferova et al., 2011 and 2013). Furthermore, activin inhibited proliferation of tumor-suppressive epidermal gd T cells, resulting in their progressive loss during tumor promotion. It also increased the number of macrophages in pre-tumorigenic skin lesions and promoted their differentiation into a phenotype resembling tumor-associated macrophages. This is functionally relevant, since depletion of macrophages reduced activin-induced skin carcinogenesis (Antsiferova et al., 2017). Finally, activin A overexpression strongly promoted the reprogramming of normal skin fibroblasts into cancer-associated fibroblasts (CAFs) through a novel activin A-mDia2-MIRO1 signaling axis (Cangkrama et al., 2020a, Cangkrama et al., 2022). Inhibition of this axis at multiple levels affected mitochondrial positioning and function, secretion of pro-tumorigenic proteins, and ultimately skin tumor formation in mouse models. The human relevance of these results is reflected by the strongly increased expression of activin A in cutaneous basal and squamous cell carcinomas and already in some skin cancer precursor lesions and by the negative correlation between activin A/mDia2/MIRO1 expression with survival in different types of human cancers (Antsiferova et al., 2011; Cangkrama et al., 2020a, Cangkrama et al., 2022). These findings highlight the parallels between wound healing and cancer and suggest inhibition of activin action as a promising strategy for the treatment of cancers overexpressing this factor (Cangkrama et al., 2020b).
When we analyzed the effect of activin A on mitochondria, we made the interesting discovery that different types of epithelial cancer cells transfer their mitochondria into adjacent fibroblasts, thereby inducing a pro-tumorigenic CAF phenotype. These results identify a key role of mitochondrial transfer in CAF differentiation (Cangkrama et al., 2025).
In addition to the skin, we demonstrated an important role of activin in other types of inflammatory and repair processes (reviewed by Werner and Alzheimer, 2006). Collaborative studies with the laboratory of Prof. Christian Alzheimer at the University of Erlangen-Nürnberg, Germany identified an important role of activin in neuroprotection, synaptic plasticity, anxiety and depression (Tretter et al., 2000; Müller et al., 2006, Tseng et al., 2008, Link et al., 2016).
Current co-workers:
- Lea Adams () (from Germany)
- Dr. Liliana Bento Lopes () (from Portugal)
- Dr. Luca Ferrarese () (from Italy)
- Joanne Gerber () (from Switzerland)
- Dr. Ismail Kücükaylak () (from Turkey)
- Huan Liu () (from China)
- Dr. Sebastian Meurant () (from Belgium)
- Dr. Deborah Stefanova () (from Bulgaria)
- Dr. Corinne Urwyler () (from Switzerland)
- Dr. Till Wüstemann () (from Germany)
- Xiaoyu Wu () (from China)
Former co-workers:
- Karin Angermeyer (from Germany)
- Dr. Maria Antsiferova () (from Russia)
- Dr. Marc Bachofner () (from Switzerland)
- Dr. Casimir Bamberger () (from Germany)
- PD Dr. Hans-Dietmar Beer () (from Germany)
- Dr. Tobias Beyer () (from Switzerland)
- Katharina Blatter () (from Switzerland)
- Dr. Kerstin Bleuel () (from Germany)
- Dr. Katharina Birkner () (from Germany)
- Dr. Friederike Böhm () (from Germany)
- Dr. Mattia Bordoli () (from Switzerland)
- Dr. Maria Brauchle () (from Germany)
- Dr. Susanne Braun () (from Germany)
- Dr. Salome Brütsch () (from Switzerland)
- Dr. Philippe Bugnon () (from Switzerland)
- Dr. Michael Cangkrama () (from Indonesia)
- Dr. Johanna Dammeier () (from Germany)
- Irene Dick (from Germany)
- Silke Durka () (from Germany)
- Dr. Felix Engelhardt (from Germany): Felix Engelhardt:
- Dr. Abbie Fearon () (from England)
- Prof. Dr. Stefan Frank () (from Germany)
- Prof. Dr. Richard Grose () (from England)
- Dr. Marcus Gassmann () (from Germany)
- Dr. Eric Haertel () (from Germany)
- Dr. Tobias Heatta-Speicher () (from Germany)
- Dr. Moritz Hertel () (from Germany)
- Dr. Griseldis Hübner-Kroll () (from Germany)
- Katharina Huggel (from Switzerland)
- Dr. Susanne Kaesler () (from Germany)
- Dr. Heidi Kögel (from Germany)
- Dr. Katalin Romancuk (Korodi) () (from Hungary)
- Dr. Monika Krampert () (from Germany)
- Dr. Andrii Kuklin ()
- Dr. Luigi Maddaluno () (from Italy)
- Dr. Marianne Madlener (from Germany)
- Dr. Michael Meyer () (from Switzerland/South Africa)
- Michelle Meyer (from Switzerland)
- Dr. Anna-Katharina Müller Bar () (from Switzerland)
- Dr. Mischa Müller () (from Switzerland)
- Dr. Christine Munding (from Germany)
- Prof. Dr. Barbara Munz () (from Germany)
- Prof. Dr. Khondokar Nasirujjaman () (from Bangladesh)
- Dr. Susagna Padrissa-Altes () (from Spain)
- Dr. Sandra Pankow () (from Germany)
- Dr. Sophia Pantasis () (from Switzerland)
- Dr. Aleksandra Piwko-Czuchra () (from Poland)
- Dr. Tamara Ramadan (from Croatia) ()
- Dr. Theresa Rauschendorfer () (from Germany)
- Helga Riesemann (from Germany)
- Dr. Diana Rotzer (from Germany)
- Dr. Coenraad Slabber () (from South Africa)
- Dr. Andreas Stanzel (from Germany)
- Dr. Heike Steiling () (from Germany)
- Dr. Jitka Somandin (Sulcova) () (from Czech Republic)
- Dr. Irmgard Thorey (from Germany)
- Dr. Laurence Vindevoghel () (from France)
- Dr. Elina Virolainen (from Finland)
- Dr. Miriam Wankell () (from Germany)
- Frédérique Wanninger (from Germany)
- Dr. Silke Sulyok (Werner) () (from Germany)
- Prof. Dr. Mateusz Wietecha () (from USA/Poland)
- Daria Wüst () (from Switzerland)
- Dr. Shen Yan () (from China)
- Prof. Dr. Jingxuan Yang () (from China)
Project 2:
Role of stress-regulated transcription factors in tissue repair, inflammatory disease and cancer (currently funded by ETH Zürich (Open ETH Project SKINTEGRITY.CH and collaborative research grant), Swiss National Science Foundation); previously funded by AETAS Foundation, Boehringer Ingelheim Fonds, Deutsche Forschungsgemeinschaft, EMBO, European Union, Hochschulmedizin Zürich, Innosuisse, Leopoldina, Promedica Foundation, CE.R.I.E.S. Award, Government of Canada, Helmut Horten Foundation, Gebert-Rüf Foundation, KTI, Wilhelm Sander Foundation, Studienstiftung des Deutschen Volkes, Jasso Graduate Scholarship, Japan).
In our search for FGF7-regulated genes, we identified the gene encoding the NRF2 transcription factor. NRF2 is a key regulator of the cellular stress response, since it regulates the expression of various cytoprotective proteins, including enzymes that detoxify reactive oxygen species. The analysis of NRF2 function in tissue repair and cancer is a major focus of the research in our laboratory.
We demonstrated that NRF2 expression is regulated by FGF7 (Braun et al., 2002) and activated in keratinocytes in response to electrophilic chemicals (Durchdewald et al., 2007). Functional studies revealed that Nrf2-deficiency in mice results in prolonged wound inflammation (Braun et al., 2002), although Nrf2 in myeloid cells is dispensable for wound repair (Joshi and Werner, 2017). We further found that Nrf transcription factors in keratinocytes are essential for skin tumor prevention (auf dem Keller et al., 2006). To determine the consequences of Nrf2 activation in the skin, we generated transgenic mice expressing a constitutively active Nrf2 mutant in keratinocytes. Activation of Nrf2 target genes strongly reduced UVB cytotoxicity through enhancement of ROS-detoxification. Remarkably, the protective effect was extended to neighbouring cells. Using different combinations of genetically modified mice we demonstrated that Nrf2 activates the production, recycling and release of glutathione and cysteine by suprabasal keratinocytes, resulting in protection of basal cells in a paracrine, glutathione/cysteine-dependent manner. These results identify Nrf2 as a key regulator in the UV response of the skin (Schäfer et al., 2010; Schäfer and Werner, 2015). However, we also showed that prolonged and excessive Nrf2 activation in keratinocytes is deleterious and results in development of an ichthyosis-like skin disease that is characterized by epidermal hyperplasia, hyperkeratosis and a defect in the epidermal barrier (Schäfer et al., 2012). This barrier function defect is likely to be further promoted by defective desmosomes. These resulted from Nrf2-mediated upregulation of microRNA-29, which targets the desmosomal component desmocollin-2 in keratinocytes (Kurinna et al., 2014). Interestingly, these studies also revealed that Nrf2 links antioxidant defense with epidermal barrier function (Schäfer et al., 2012) and with inflammatory processes (Kurinna/Muzumdar et al., 2016). We also identified an unexpected function of activated Nrf2 in the pathogenesis of chloracne (MADISH), a skin pathology that develops upon exposure to dioxin or related toxins (Schäfer et al., 2014). On the other hand, we found that Nrf2 activation ameliorates the phenotype in a mouse model of the severe genetic skin disease Netherton Syndrome (Muzumdar et al., 2020). Very recently, we showed in a quantitative proteomics approach that NRF2 activity is reduced in the epidermis of patients with Atopic Dermatitis, suggesting that limited activation of NRF2 may also have beneficial effects in this major inflammatory skin disease (Koch et al., 2022 and 2024). Unfortunately, however, even mild chronic activation of Nrf2 in keratinocytes can be deleterious if the skin is affected by oncogenic mutations. Thus, chronic activation of Nrf2 in keratinocytes caused a severe increase in tumor incidence and multiplicity in a transgenic mouse model of virus-induced skin tumorigenesis (Rolfs et al., 2015). Activation of Nrf2 in mouse fibroblasts unexpectedly induced senescence of these cells, and the resulting senescence-associated secretory phenotype strongly promoted wound repair. This resulted from Nrf2-mediated regulation of different matrix proteins and deposition of a senescence-promoting matrisome. However, the same mechanism also promoted skin cancer formation in xenograft models, revealing the bright and the dark sides of Nrf2 under different situations (Hiebert et al., 2018). The results also reveal an unexpected pro-tumorigenic activity of Nrf2 in the skin. Overexpression/activation of Nrf2 in mouse fibroblasts also suppressed the expression of major collagens through induction of Nrf2-regulated miRNAs, resulting in severe skin weakening as seen in aged murine and human skin (Hiebert et al., 2022). These activities should be considered when NRF2 activating compounds are used for skin protection under stress conditions. In a search for other transcription factors, which cooperate with NRF2 in the skin, we showed that NRF2 interacts with p63 at promoters and enhancers in human keratinocytes. The combined activity of both transcription factors is crucial for the maintenance of keratinocyte proliferation in the epidermis, in particular in cells that are already committed to differentiation (Kurinna et al., 2021).
In addition to its potent role in the skin, we identified a crucial function of Nrf2 in liver regeneration through its capacity to regulate insulin/insulin-like growth factor signaling in the injured liver (Beyer et al., 2008), and we also demonstrated that Nrf2 protects from toxin-induced liver fibrosis (Xu et al., 2008). In the normal liver, we discovered a collaborative function of Nrf2 and NF-kB, since combined loss of these transcription factors in hepatocytes caused severe liver inflammation and development of inflammatory hepatocellular adenomas in mice (Köhler et al., 2015; Kuklin et al., 2025). However, further activation of Nrf2 in the regenerating liver is also deleterious, since activated Nrf2 was found to regulate target genes in hepatocytes of the regenerating liver, which enhance apoptosis and inhibit proliferation (Köhler et al., 2014).
One of the targets of Nrf2 is peroxiredoxin 6, an enzyme, which detoxifies hydrogen peroxide and organic peroxides. We demonstrated that peroxiredoxin 6 is strongly expressed in the hyperproliferative epidermis of skin wounds and of psoriatic lesions (Frank et al., 1997; Munz et al., 1997). These high levels of peroxiredoxin 6 are likely to be biologically important, since overexpression of this enzyme in the epidermis of transgenic mice enhanced wound healing in aged animals and strongly protected keratinocytes from UVB toxicity (Kümin et al., 2006). In the absence of peroxiredoxin 6, UV-induced cell damage in the epidermis was strongly enhanced, confirming the important protective function of this enzyme for keratinocytes. In addition, peroxiredoxin 6 was found to be crucial for blood vessel integrity in wounded skin (Kümin et al., 2007). We also identified an important dual function of peroxiredoxin 6 in skin carcinogenesis: protection from skin tumor development, but enhancement of the malignant progression of existing tumors (Rolfs et al., 2013). Another target of Nrf2 is Gclc, the rate-limiting enzyme in the biosynthesis of the antioxidant glutathione. We showed that a glutathione-Nrf2-thioredoxin cross-talk ensures keratinocyte survival and efficient wound repair (Telorack et al., 2016).
In contrast to the well-characterized Nrf2, little is known about the biological functions and mechanisms of action of the related Nrf3 transcription factor. We identified Nrf3 as an important regulator of the UV response of the skin. Thus, Nrf3 promotes UV-induced keratinocyte apoptosis through the regulation of cell-cell and in particular cell-matrix adhesion (Siegenthaler/Defila et al., 2018). Our recent work identified NRF3 as a potent tumor suppressor in the skin through its capacity to regulate the unfolded protein response (Gurri et al., 2023).
In other ongoing work we investigate the role of transcription factors, which are activated by hyperosmotic or mechanical stress in different skin cells and their downstream targets and biological effects. We showed that hyperosmotic stress in keratinocytes, which results from a defect in the epidermal barrier and the resulting water loss and skin dryness, induces the expression of various genes/proteins that protect the epidermis from hyperosmotic stress, including the cell-cell adhesion protein CLCA2 (Seltmann/Meyer et al., 2018; Seltmann 2024). Hyperosmotic stress also occurs in dermal fibroblasts upon skin wounding. We identified serine proteinase 35 (PRSS35) as an osmo-regulated gene/protein, which is upregulated in healing skin wounds. This is functionally important, because PRSS35 regulates the matrisome in response to hyperosmotic stress, thereby controlling cell proliferation (Sänger et al., 2023).
Current co-workers:
- Odysseus Grünert () (from Austria)
- Fumimasa Kubo () (from Japan)
- Dr. Kristin Seltmann () (from Germany)
- Dominik Zanetti () (from Switzerland)
Former co-workers:
- Prof. Dr. Ulrich auf dem Keller (from Germany) (deceased 2023)
https://onlinelibrary.wiley.com/doi/10.1111/wrr.13118
- Nadja Bain () (from Switzerland)
- Dr. Maya Ben-Yehuda Greenwald () (from Israel)
- Dr. Tobias Beyer () (from Switzerland)
- Christiane Born-Berclaz () (from Switzerland)
- Dr. Susanne Braun () (from Germany)
- Dr. Philippe Bugnon () (from Switzerland)
- Dr. Claudia Defila () (from Switzerland)
- Dr. Sabine Dütsch () (from Switzerland)
- Dr. Nikolas Epp (from Germany)
- Dr. Selina Gurri () (from Switzerland)
- Maria Gysi (from Switzerland) ()
- Nicole Hallschmid (from Germany)
- Hayley Hiebert (from Canada)
- Dr. Paul Hiebert () (from Canada)
- Dr. Christine Huber (Hanselmann) () (from Germany)
- Dr. Natasha Joshi () (from India)
- Dr. Ulrike Köhler () (from Germany)
- Dr. Michael Koch () (from Germany)
- Dr. Andrii Kuklin () (from Ukraine)
- Dr. Svitlana Kurinna () (from Ukraine)
- Dr. Marcus Gassmann () (from Germany)
- Dr. Heidi Kögel (from Germany) ()
- Dr. Angelika Lengweiler (Kümin) () (from Switzerland)
- Dr. Franziska Metzler (Lieder) () (from Germany)
- Dr. Sukalp Muzumdar () (from India)
- Rin Okumura () (from Japan)
- Helga Riesemann (from Germany)
- Dr. Frank Rolfs () (from Germany)
- Dr. Christina Rycken (Siemes (from Germany) (deceased 2025)
- Dr. Catharina Sänger () (from Germany)
- PD Dr. Matthias Schäfer () (from Germany)
- Dr. Beat Siegenthaler () (from Switzerland)
- Dr. Michèle Telorack () (from Germany)
- Dr. Baijayanti Jha () (from India)
- Dr. Weihua Xu (from China)
- Maria Zubair () (from England)